上海人工智能实验室(上海AI实验室)、复旦大学、上海交通大学医学院附属瑞金医院及上海市病毒研究院联合团队推出 ViraHInter —— 一款融合蛋白序列与结构双模态的AI预测模型,不用开展湿实验就能预判出病毒将“劫持”哪些人体蛋白。将抗病毒药物研发从逐个病毒应对的模式,推进到针对多病毒共性宿主因子的系统化靶点发现模式。
关键成果:
- 在严苛的人类蛋白基准测试中,预测精度(AUPR 0.44)全面超越AlphaFold 3(0.23)、RoseTTAFold2-PPI(0.28)、RF2-Lite(0.10);
- 面对病毒-人类蛋白相互作用时,模型精度高达0.50,比AlphaFold 3高4.5倍 (0.11),比其他方法高6倍以上;
- 锁定33个跨流感亚型共享的宿主因子,为广谱抗流感药物研发提供全新靶点。
根据介绍,传统方法预测蛋白相互作用,要么只分析氨基酸序列(病毒的遗传密码),要么只分析三维结构(病毒的立体形态)。ViraHInter的突破在于——让AI同时精准掌握这两种信息:
- 结构分支:生成病毒-宿主蛋白复合物的全原子三维结构,精准刻画界面上每一个原子如何贴合,为药物设计打下基础;
- 序列分支:借助蛋白语言模型,从海量蛋白进化信息中识别保守模式,即使病毒快速变异也能抓住不变的模式。、
两种信息通过注意力机制深度融合——面对结构清晰的蛋白,模型更依赖结构信息;面对快速变异或无序的病毒蛋白,则更依赖序列信息。这种自适应融合,正是ViraHInter跨病毒家族泛化能力的关键。
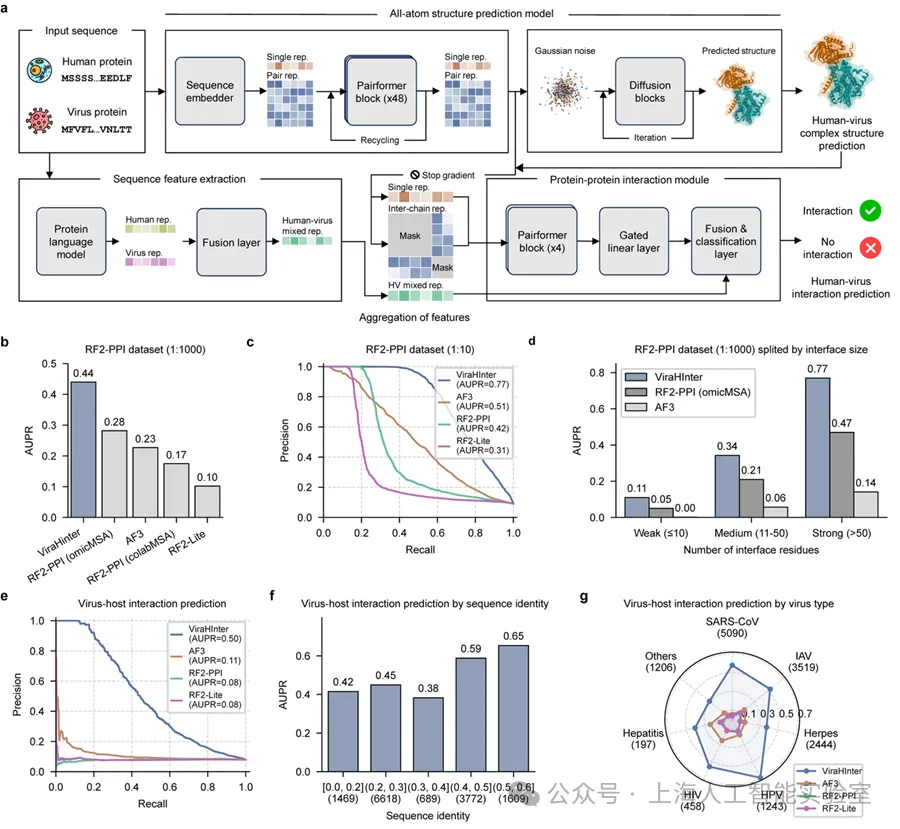
更关键的考验在于面对“陌生”病毒的能力——这直接关系到模型是否适用于新发病原体。在序列同源性严格受控的测试中(即测试病毒与训练集中病毒的序列相似度不超过60%),ViraHInter的AUPR达到0.50,比 AlphaFold 3(0.11)高4.5倍,比其他方法高6倍以上。
值得注意的是,这一差距甚至超过了常规测试下的优势,说明当训练数据中缺乏同类病毒参考时,ViraHInter的双模态架构优势更为突出——这恰恰是应对新发病原体时最需要的能力。
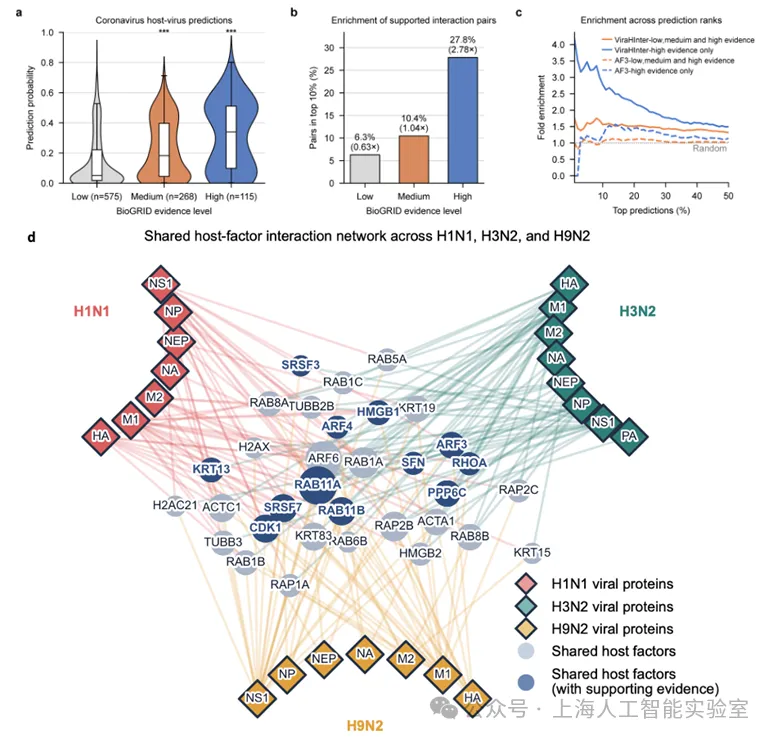
科研团队利用ViraHInter,在人类全蛋白组范围内对 H1N1、H3N2、H9N2 这三种流感亚型的10个关键病毒蛋白(包括PB2、NS1、NP、HA、PA等)进行了系统筛选,最终锁定33个被三种亚型共同靶向的核心宿主因子。其中:
- 12个因子在已有研究中已被证实参与流感感染过程,但从未在任何病毒-宿主互作数据库中被记录,反映出ViraHInter能够识别传统实验方法难以捕捉的相互作用;
- RAB11A蛋白是三种亚型血凝素共同的结合伙伴,且结合模式高度一致。针对 RAB11A与血凝素相互作用界面开发的药物,有望对多种流感亚型同时起效。
另外,在冠状病毒研究中,ViraHInter也发现了类似规律:针对SARS-CoV-1、SARS-CoV-2和MERS-CoV的分析显示,RAB8A蛋白与病毒非结构蛋白NSP7的结合界面在三种冠状病毒中高度保守,提示RAB8A可能是多种冠状病毒共同依赖的关键宿主因子,可作为广谱抗冠状病毒药物研发的靶点。
两种信息通过注意力机制深度融合——面对结构清晰的蛋白,模型更依赖结构信息;面对快速变异或无序的病毒蛋白,则更依赖序列信息。这种自适应融合,正是ViraHInter跨病毒家族泛化能力的关键。
